D1SoftballNews
Home
News
Entertainment
Business
Sports
Games
Technology
Health
Technology
Admin
April 18, 2024
0
0
They are creating a wonderful material that can replace graphene. This is a thin two-dimensional layer of gold.
Sports
Admin
April 18, 2024
0
3
Madrid asks to postpone visit to Real Sociedad to Friday
Entertainment
Admin
April 18, 2024
0
33
Justin Bieber, Jaden Smith and Noah Beck in the arms of friends and Twitter in the fund, we will meet in 2024
News
Admin
April 18, 2024
0
31
Breaking news on Iran attacks on Israel, war with Hamas and situation in Gaza, LIVE: News, reactions and more
Health
Admin
April 18, 2024
0
31
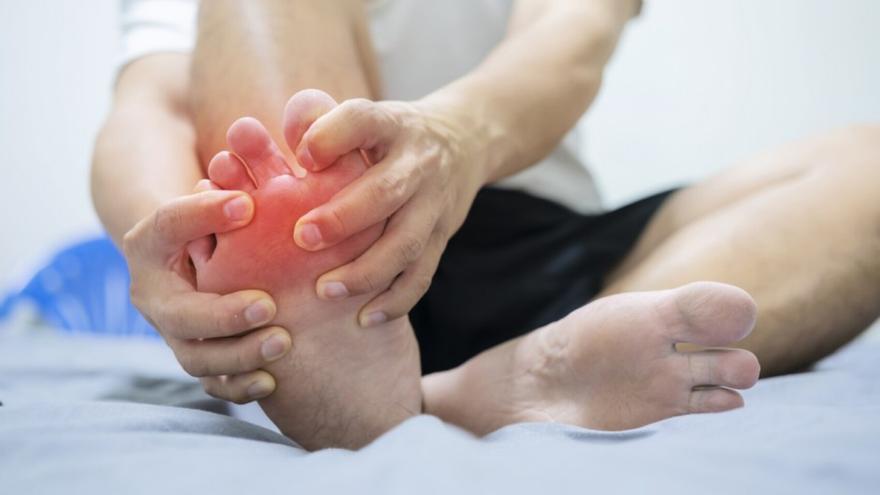
REDUCING URIC ACID: How to Lower Uric Acid and Avoid Joint Problems: Try This Food!
Business
Admin
April 18, 2024
0
39
The government is considering requiring Taqa to sell 10% of Naturgy following a takeover bid.
Technology
Admin
April 18, 2024
0
36
Jupiter’s moons have had active volcanoes since their inception – DW – 04/18/2024
Sports
Admin
April 18, 2024
0
36
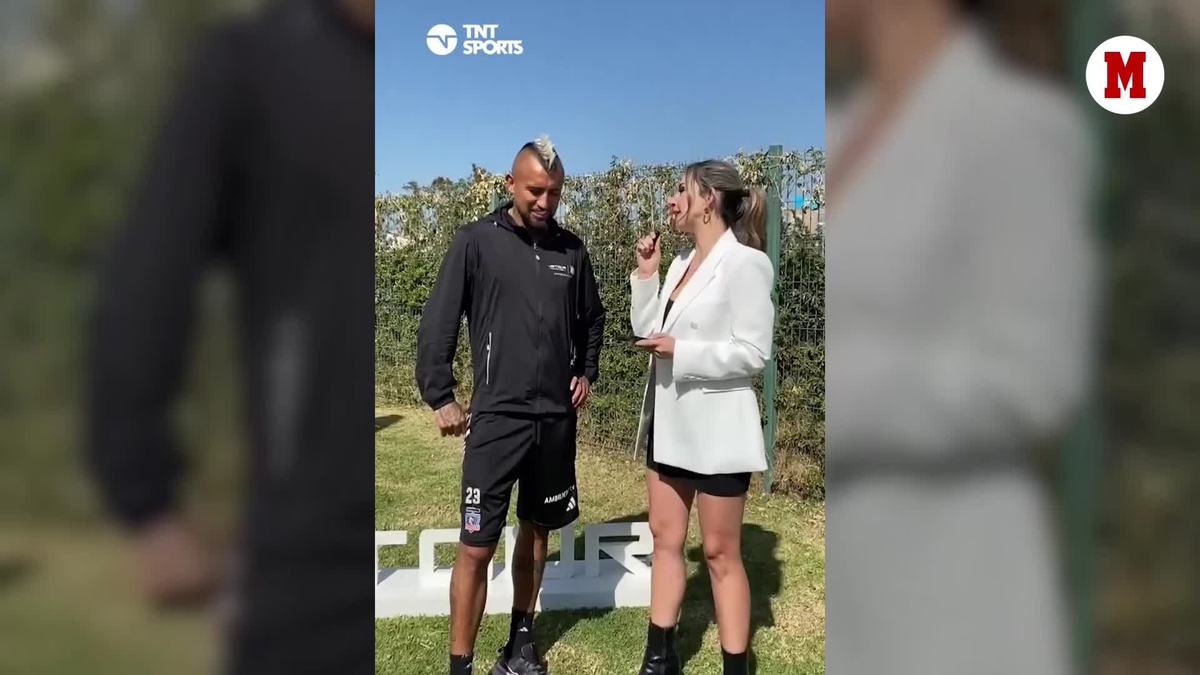
“I would like to play for Madrid”
Entertainment
Admin
April 18, 2024
0
35
When a Relationship Rumor Goes Viral
News
Admin
April 18, 2024
0
35
Trump juror apologizes for feeling intimidated
Next page
Back to top button